A progressive disease which affects upper and lower motor neurons in the brain, brainstem and spinal cord.
Pathophysiology
Although there is some evidence of a genetic or familial component, cases are largely sporadic. Various theories exist regarding the aetiology of MND, including glutamate toxicity and oxidative stress as the cause for sporadic disease.
Clinical Features
Symptoms usually begin in a focal part and will then progress to become widespread. For example, patients may present with weakness of a leg e.g. foot drop or trouble with manual dexterity. This then progresses to involve more muscles. Alternatively, onset may begin with bulbar involvement, thus presenting with dysarthria and dysphagia followed by limb involvement. Patients can also suffer from painful muscle spasms and drooling.
There are 4 main subclassifications of motor neuron disease, each of which have slightly varied presentations.
Amyotrophic Lateral Sclerosis
- This is the most common type of MND.
- Both upper motor neuron (UMN) and lower motor neuron signs (LMN)
- UMN Signs: Hyperreflexia, spasticity, weakness, positive Babinski
- LMN Signs: Denervation atrophy, weakness and fasciculations
- Other: Absent sensory signs, no autonomic involvement, no eye involvement.
- Patients may develop frontotemporal dementia
Progressive Bulbar Palsy
- Onset begins by affecting bulbar muscles, followed by the extremities. Occurs when cranial nerves IX-XII (9-12) are affected.
- Dysarthria
- Dysphagia
- Fasciculations and wasting of the tongue
- Pseudobulbar affect: Episodes of uncontrollable crying or laughter which are often inappropriate to the situation
Progressive Muscular Atrophy
- LMN variant only, so no UMN signs
- Tends to affect distal muscles before proximal
Primary Lateral Sclerosis
- Slower progressing and has a better prognosis.
- Begins with only UMN signs, with LMN eventually becoming part of the clinical picture.
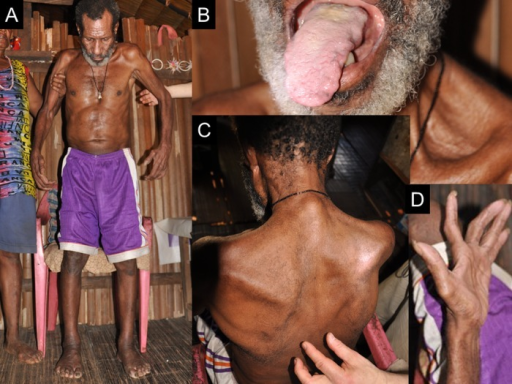
Okumiya K, Wada T, Fujisawa M, Ishine M, Garcia Del Saz E, Hirata Y, Kuzuhara S, Kokubo Y, Seguchi H, Sakamoto R, Manuaba I, Watofa P, Rantetampang AL, Matsubayashi K - BMJ Open (2014), CC BY 3.0 , via Wikimedia Commons
Image A shows an inability to stand without support, B-D show atrophy
Investigations
MND is largely a clinical diagnosis, making the history and examination an important aspect of diagnosis. Investigations that may be conducted include:
- Electromyography (EMG)
- Nerve stimulation tests
- MRI brain and spine: To rule out nerve compression
- Vitamin B12: Rule out peripheral neuropathy
- Genetic testing: Particularly useful if familiar motor neuron disease is suspected
Management
MND is a progressive condition, and prognosis is poor. Median survival is 3 to 5 years. Management is largely supportive.
- Riluzole: A glutamate release antagonist which can slow the progression of the disease by a few months.
- Muscle relaxants to treat spasticity such as baclofen
- Speech and language therapy for dysarthria
- Percutaneous endoscopic gastrostomy (PEG Tube): Due to dysphagia
- Ventilation: Patients with MND can have weakness of the respiratory muscles, thus potentially necessitating either non-invasive or invasive ventilation
References
https://neuropathology-web.org/chapter9/chapter9fALS.html
https://www.ncbi.nlm.nih.gov/books/NBK556151/