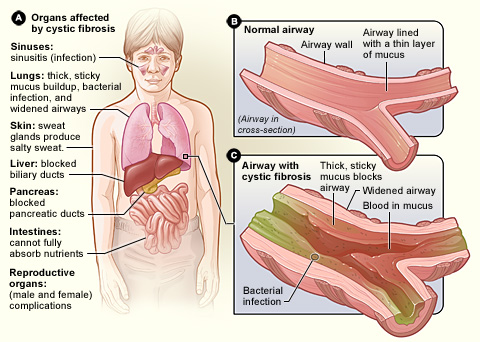
Cystic fibrosis is an autosomal recessive condition that affects bodily mucus secretion. The lungs are primarily affected although other organs such as the pancreas and digestive tract can also be involved.
Pathophysiology
- The cystic fibrosis transmembrane protein regulator (CFTR) is a protein found on epithelial cells and it is responsible for pumping chloride ions into various secretions such as mucus, sweat and pancreatic fluid.
- This secretion of chloride ions also draws sodium ions through passive sodium channels leading to high quantities of sodium and chloride ions in secretions which will draw water and thin out secretions.
- Cystic fibrosis occurs when there is a mutation in the cystic fibrosis (CF) gene found on chromosome 7, which codes for the CFTR protein. The most common mutation is the delta F508 mutation.
- This mutation results in reduced chloride (and by extension, sodium) secretion into mucus. This results in extremely thick and viscous mucus due to reduced water reabsorption.
- Due to hyperviscous secretions, mucus plugs obstruct the smaller ducts within organs – this stasis of e.g. mucus leads to infection and inflammation which ultimately results in organ damage over time.
National Heart Lung and Blood Institute (NIH), Public domain, via Wikimedia Commons
Clinical Features of Cystic Fibrosis
Location of CFTR
Anywhere the CFTR protein is present can be affected by cystic fibrosis. This includes:
- Respiratory Tract (lungs, sinuses)
- Pancreas
- Liver
- Digestive tract
- Sebaceous and Eccrine sweat glands
Clinical Features
Lungs
The lungs are normal in utero and at birth. With time, as the mucus becomes thicker, infections begin to develop within the lungs which results in inflammation. This usually leads to bronchiectasis in childhood. Repeated episodes of bronchiectasis ultimately result in lung damage.
- Cough, which is usually productive
- Recurrent chest infections
- Nasal polyps
- Dyspnoea
- Sinusitis
Pancreas
Thick mucus plugs are able to block the pancreatic ductal system leading to a deficiency in pancreatic zymogens reaching the duodenum resulting in malabsorption and steatorrhea. Thus, children with cystic fibrosis might present with failure to thrive. Patients often have co-existing fat-soluble vitamin deficiencies (A, D, E and K). Damage to the pancreas can lead to diabetes mellitus.
Hepatobiliary
The biliary ductal system may become blocked with thick secretions which can lead to obstructive jaundice, cirrhosis, portal hypertension and even splenomegaly.
Gastrointestinal Tract
Meconium ileus may occur in children which is where lack of hydration of bowel contents leads to a mechanical obstruction. Bowel obstruction can also occur later in life for the same reason. It is also possible to develop rectal prolapse due to cystic fibrosis.
Reproductive Tract
It is thought that 97-98% of men with cystic fibrosis are infertile due to congenital bilateral absence of the vas deferens. Women can have thicker cervical mucus which may make it difficult to get pregnant, but they are not infertile.
Investigations
Special Tests
- Sweat Test: Ordinarily, the CFTR channel is responsible for pumping chloride into various different secretions. In the sweat glands, however, the channel aims to pump chloride ions from the lumen into sweat gland cells i.e. reabsorb chloride ions (and subsequently sodium). Thus, in cystic fibrosis there is a lack of reabsorption of chloride ions (and sodium ions) leading to high levels of sodium chloride in the sweat. Chloride ion levels in excess of 60mmol/L suggests a diagnosis of cystic fibrosis.
- Genetic Testing: can be used to confirm the diagnosis by screening for mutations in the CF gene.
Bedside
- Sputum culture: Look for lower respiratory tract infection
- Oral glucose tolerance test (diabetes): Pancreatic destruction
- Faecal elastase test: Check for pancreatic insufficiency
Bloods
- FBC: Patients with cystic fibrosis may have anaemia
- Clotting tests: Vitamin K function
- Liver function tests: Hepatobiliary effects
- Vitamin A, D, E: Due to lack of pancreatic zymogens
Imaging
- Chest x-ray: Rule out other causes
- Abdominal ultrasound: Hepatobiliary effects
Management
Cystic fibrosis cannot be cured, but it is possible to manage the symptoms. A multi-disciplinary approach should be used i.e. alongside the respiratory consultant and GP, a physiotherapist and dietician can also be involved.
Routine Reviews
Patients with CF should receive regular reviews e.g. 3-6 monthly to monitor things like their pulmonary function, the presence of diabetes, osteoporosis, liver disease, vitamin D deficiency etc.
Infection Management
- Patients with cystic fibrosis can be infected by various different organisms including staphylococcus aureus, pseudomonas aeruginosa, burkholderia cepacia complex, and haemophilus influenza.
- NICE recommend offering flucloxacillin as prophylaxis against staphylococcus aureus infection from diagnosis up to the age of 3, and potentially continuing this until the age of 6.
- Individuals who continue to have a deterioration in their lung function, or have repeated exacerbations, can be offered azithromycin at an immunomodulatory dose. If a deterioration in lung function/exacerbations continues despite azithromycin, oral steroids can be considered as an alternative.
Managing Chest Symptoms
- Airway clearance and postural drainage techniques: These are taught by a physiotherapist and can help clear the thick mucus plugs that develop in the lungs. For younger patients, parents are usually taught how to perform the clearance techniques.
- Mucoactive agents: These are drugs which help to clear mucus from the airways. Dornase alpha; recombinant human deoxyribonuclease (rhDNase) is the first line drug offered in the UK, as per NICE guidance with hypertonic sodium chloride being used either as an adjunct or as a second line.
Nutritional Support
- Oral pancreatic enzyme therapy e.g. Creon should be offered where there is pancreatic insufficiency – this helps to replace pancreatic enzymes which don’t quite make it to the digestive tract due to the thickness of pancreatic secretions.
- Patients with cystic fibrosis should be encouraged to eat large portions and high-energy foods in attempts to increase calorie intake.
- If conservative measures of increasing calorific intake do not work, enteral tube feeding may be necessary
References
https://www.cff.org/Life-With-CF/Transitions/Reproductive-Health-and-Fertility/CF-and-the-Male-Reproductive-System/Fertility-in-Men-With-CF/
https://www.cff.org/Life-With-CF/Transitions/Reproductive-Health-and-Fertility/CF-and-the-Female-Reproductive-System/Fertility-in-Women-With-CF/
https://web.stanford.edu/class/psych121/humangenome-CF.htm#sweat
https://www.ncbi.nlm.nih.gov/pubmed/18849562
https://www.nice.org.uk/guidance/ng78/chapter/Recommendations#pulmonary-monitoring-assessment-and-management